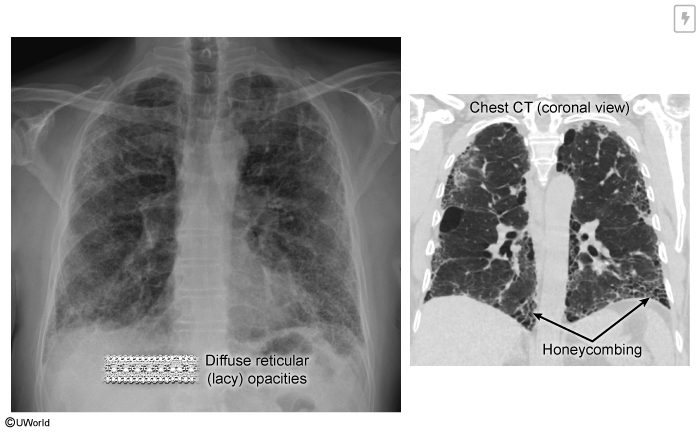
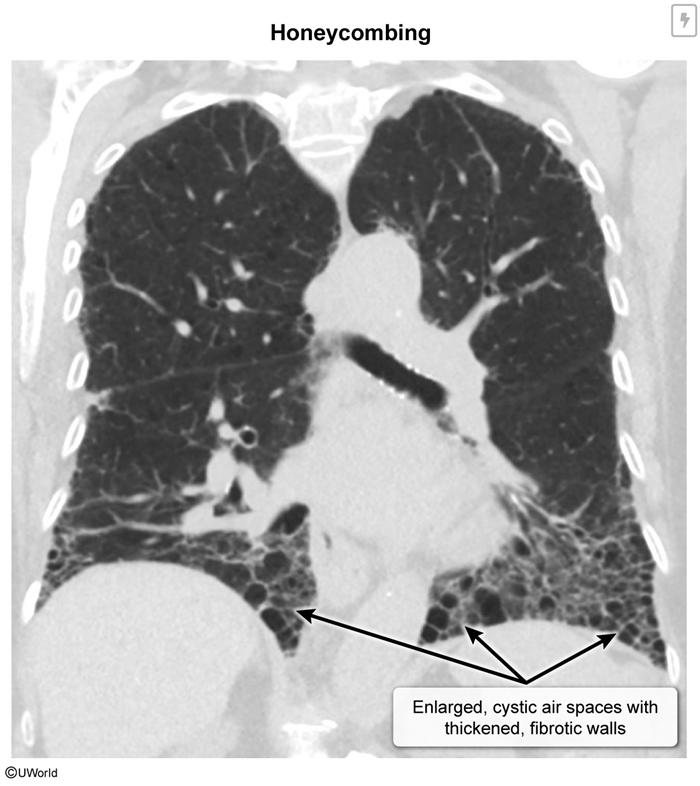
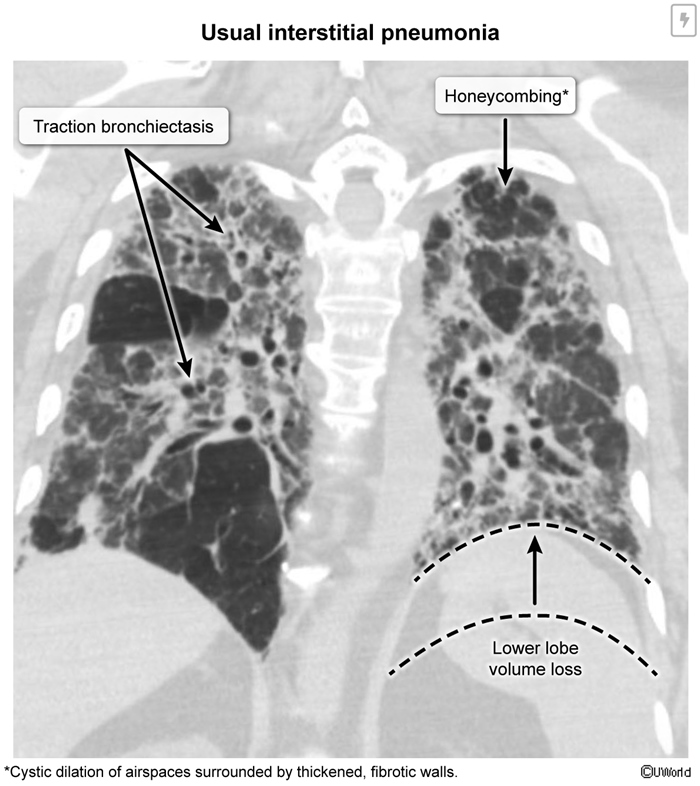
Idiopathic Pulmonary Fibrosis (IPF)
Article Sections
Introduction
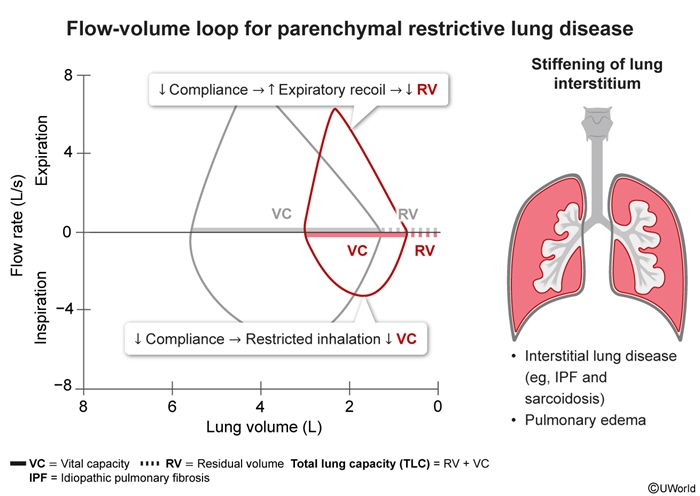
Idiopathic pulmonary fibrosis (IPF) is an interstitial lung disease (ILD) characterized by progressive fibrotic changes in the lung parenchyma leading to respiratory function decline, morbidity, and mortality. IPF affects roughly 1 in 4,000 adults worldwide. By definition, IPF lacks a definite underlying cause, although recent advances suggest genetic and environmental contributing factors.
Pathogenesis
IPF is an aberrant wound-healing response to repetitive alveolar epithelial injury. Instead of normal restoration, the alveolar surface is replaced via fibrosis. Key steps in pathogenesis include:
- Epithelial injury: IPF likely begins with an initial insult to the alveolar epithelium (eg, due to smoking, acid reflux, recurrent aspiration, or viral infections). Type I pneumocytes, the squamous epithelial cells that cover 90% of the alveolar surface, are damaged and depleted.
Continue Learning with UWorld
Get the full Idiopathic Pulmonary Fibrosis (IPF) article plus rich visuals, real-world cases, and in-depth insights from medical experts, all available through the UWorld Medical Library.
Unlock Full AccessFigures
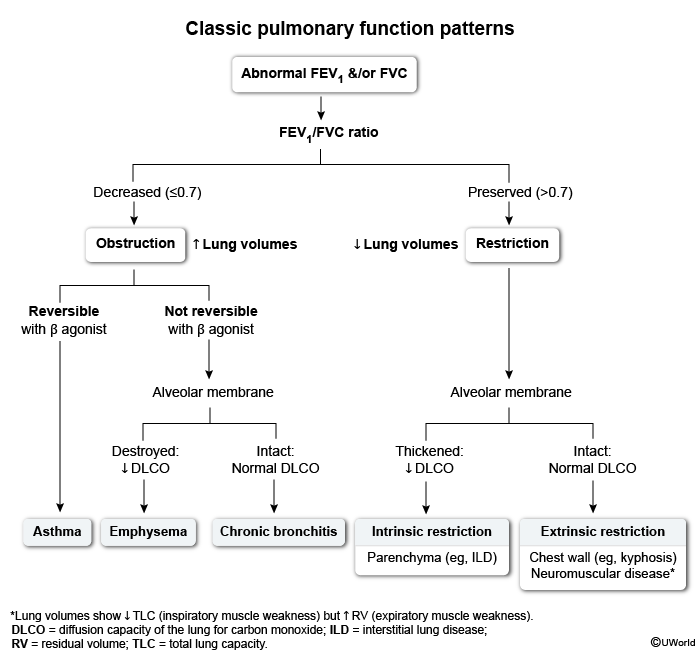
Images