Cystic Fibrosis
Article Sections
- Introduction
- Pathogenesis
- Clinical presentation
- Diagnosis
- Differential diagnosis
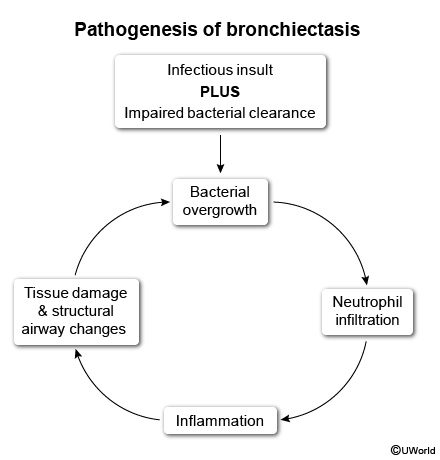
- Bronchiectasis and recurrent pulmonary infections
- Obstructive lung disease and hepatic or pancreatic disease
- Infant with delayed meconium passage
- Management of CF pulmonary disease
- Management of CF extrapulmonary disease
- CFTR modulators
- Prognosis
- Summary
Introduction
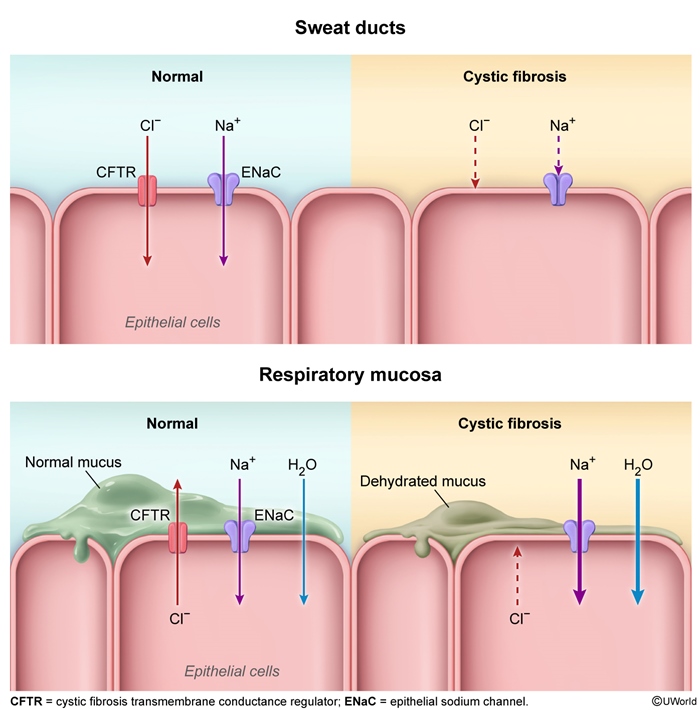
Cystic fibrosis (CF) involves production of abnormally thick epithelial mucus leading to progressive scarring and dysfunction across multiple organs, particularly in the lungs, pancreas, liver, and reproductive systems. It is the most common monogenic disorder, affecting ~1/3,000 live births, with ~1/25 Caucasian adults being carriers.
Pathogenesis
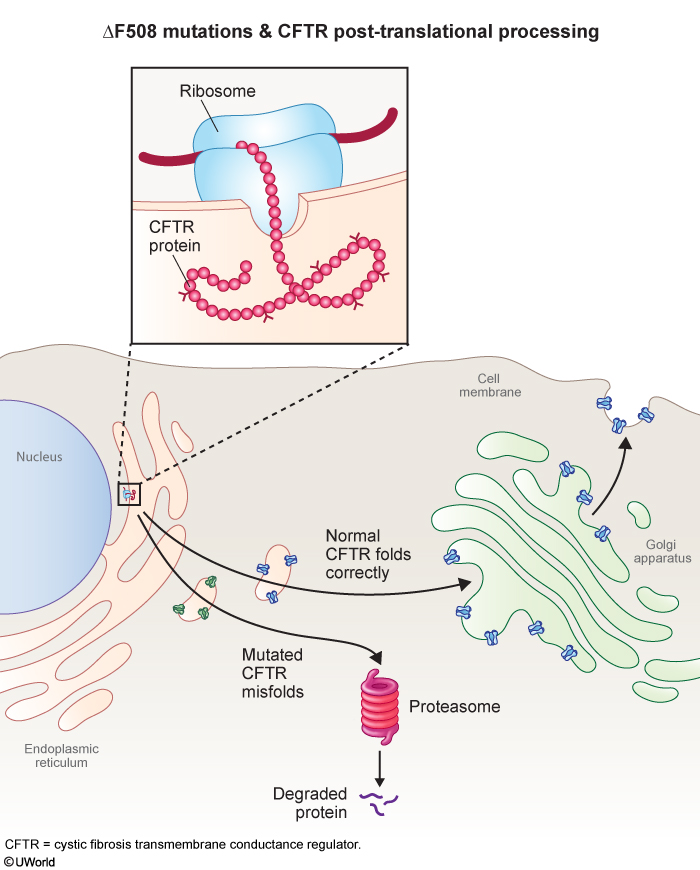
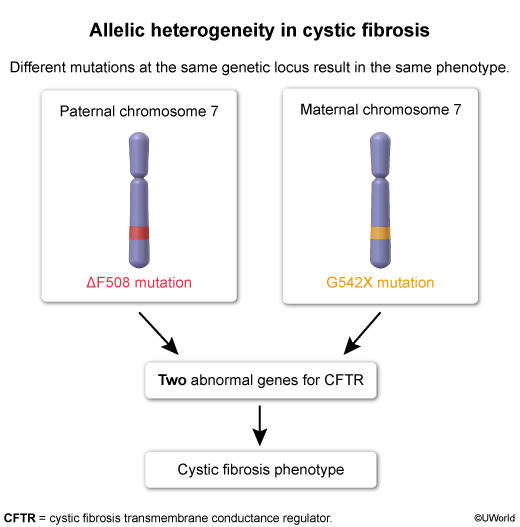
CF is an autosomal recessive disorder due to inheritance of 2 abnormal copies of the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Mutations in CFTR result in a protein that is truncated (eg, G542X), is nonfunctional (eg, G551D), or fails to reach the cell surface (ΔF508). ΔF508 is the most common mutation, accounting for ~70% of CF cases. ΔF508 (ie, deletion of phenylalanine at position 508) leads to misfolding and subsequent proteasomal degradation of the CFTR protein (
Continue Learning with UWorld
Get the full Cystic Fibrosis article plus rich visuals, real-world cases, and in-depth insights from medical experts, all available through the UWorld Medical Library.
Unlock Full AccessFigures