Huntington Disease
Article Sections
Introduction
Huntington disease is a progressive, fatal, autosomal dominant neurodegenerative disorder characterized by a triad of movement abnormalities, cognitive decline, and psychiatric disturbances.
Pathophysiology
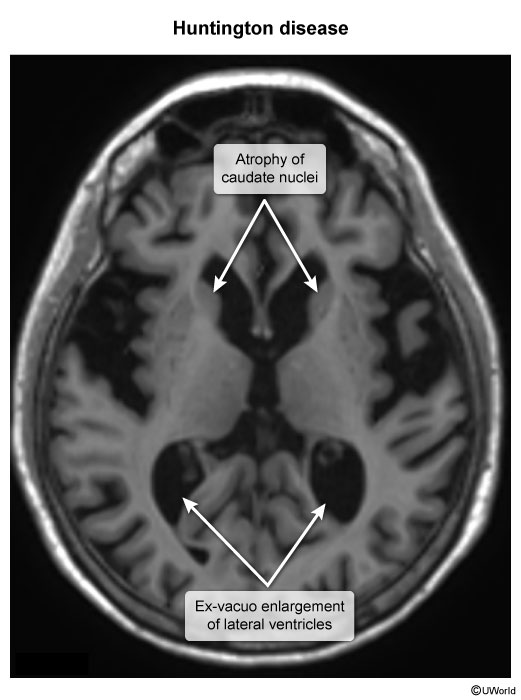
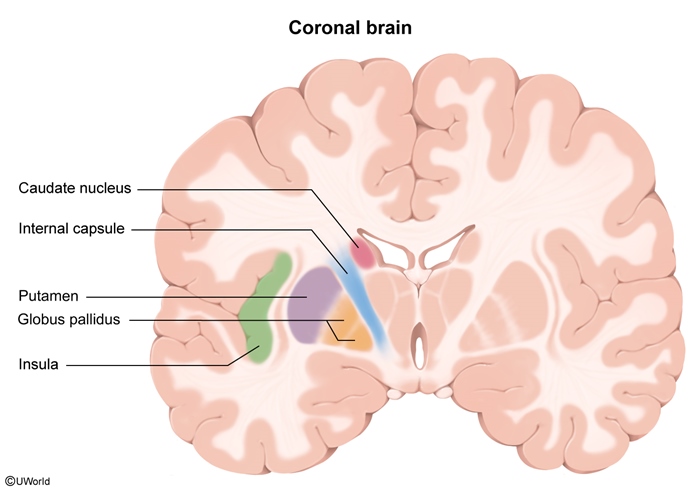
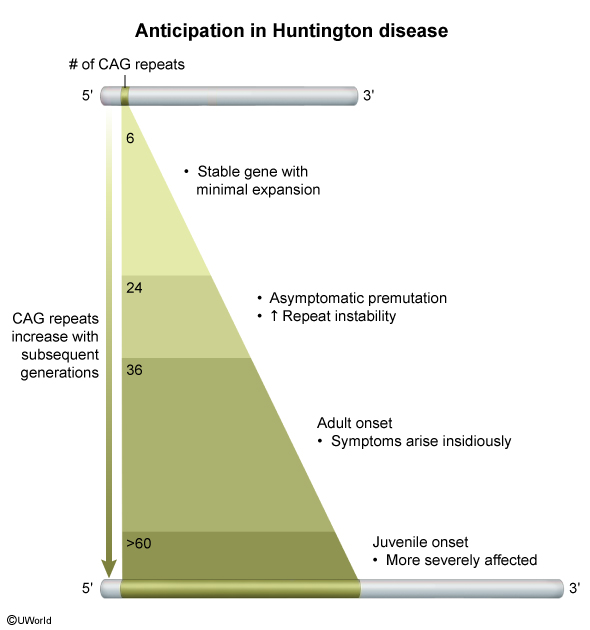
Huntington disease is caused by an autosomal dominant CAG trinucleotide repeat expansion in the huntingtin (HTT) gene on chromosome 4p. This leads to an expanded polyglutamine sequence on the huntingtin protein, which results in a gain-of-function mutation that causes toxic accumulation and pathologic interaction with other proteins, including various transcription factors. As huntingtin is widely expressed in the brain, death of inhibitory (GABA) neurons in the striatum (caudate nucleus and putamen), which help regulate the movement and behavior centers in the cortex, leads to classic features of Huntington disease, including chorea, behavioral abnormalities (eg, irritability), and progressive dementia.
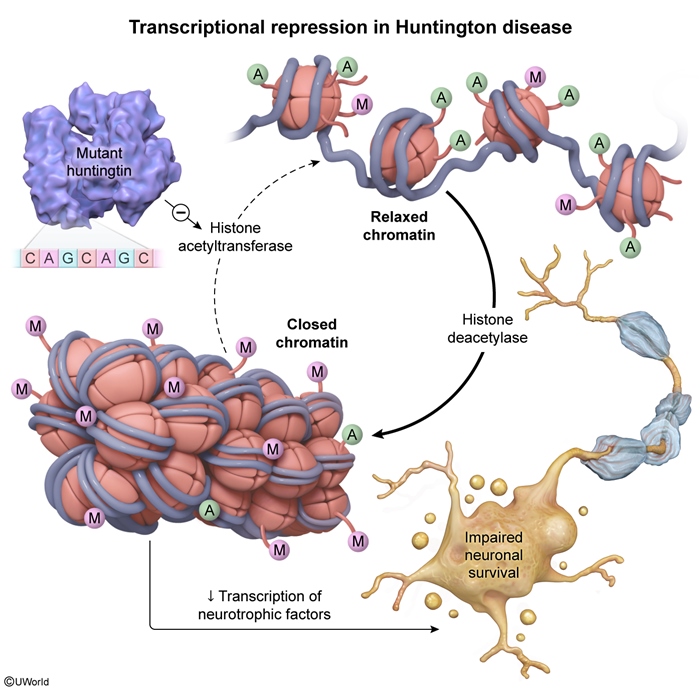
Transcriptional repression (silencing) is one of the mechanisms by which mutated huntingtin is thought to cause disease (
Continue Learning with UWorld
Get the full Huntington Disease article plus rich visuals, real-world cases, and in-depth insights from medical experts, all available through the UWorld Medical Library.
Unlock Full AccessFigures
Images