Sickle Cell Disease
Article Sections
Introduction
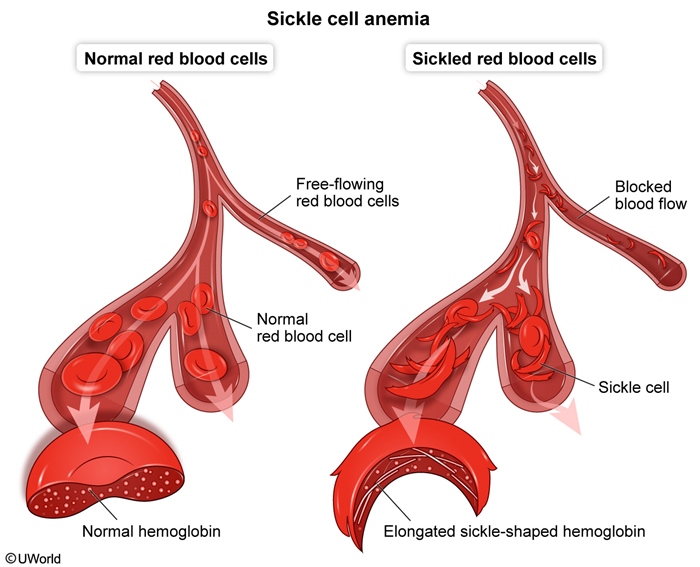
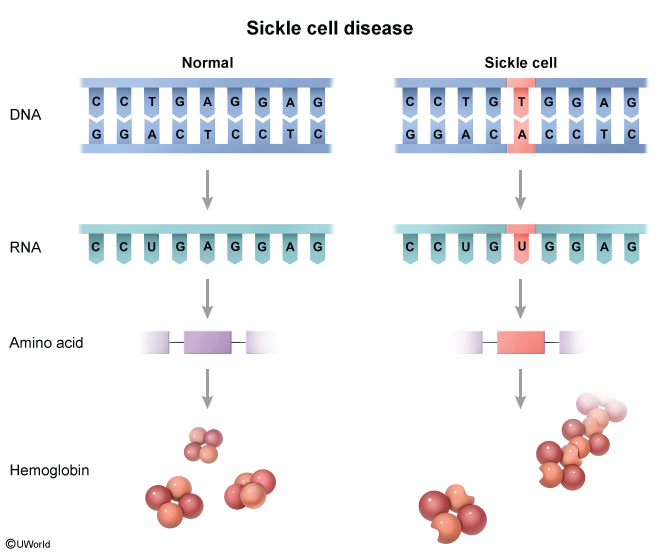
Sickle cell disease (SCD) is a group of inherited hemoglobinopathies characterized by the predominance of hemoglobin S (Hb S), a structural variant of hemoglobin, within red blood cells (RBCs). Unlike normal hemoglobin, Hb S polymerizes under hypoxic conditions, forming rigid sickle-shaped RBCs. Patients have lifelong chronic hemolysis and vaso-occlusion (Figure 1) that can cause both acute (eg, ischemic pain) and chronic (eg, chronic kidney disease) complications.
Pathogenesis
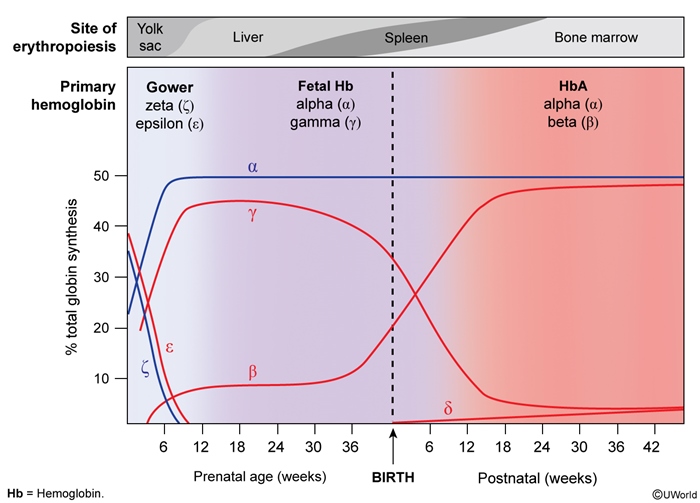
Hemoglobin is composed of 4 globin chains and carries oxygen in RBCs. Normally, 2 different pairs of globin chains (eg, a pair of alpha globin chains and a pair of beta globin chains) join to form a tetramer (Figure 2). Globin chain synthesis is a highly regulated process that results in a predominant tetramer (
Continue Learning with UWorld
Get the full Sickle Cell Disease article plus rich visuals, real-world cases, and in-depth insights from medical experts, all available through the UWorld Medical Library.
Unlock Full AccessFigures