Amyloidosis
Article Sections
Introduction
Amyloidosis is characterized by the extracellular deposition of amyloid proteins, which misfold and aggregate into insoluble fibrils. Over 30 different proteins can form amyloid deposits, and the specific protein involved determines the subtype of amyloidosis and its clinical implications. Important categories of amyloidosis include the following:
- AL (light-chain) amyloidosis is caused by monoclonal plasma cell proliferation with excessive production of abnormal immunoglobulin light chains. It is the most common systemic amyloidosis and frequently affects the heart, kidneys, liver, and peripheral nerves.
- ATTR (transthyretin) amyloidosis arises from the deposition of misfolded transthyretin protein. It can be hereditary due to mutations in the TTR gene or age-related in the wild-type form, with both forms often affecting the heart and peripheral nerves.
- AA (serum amyloid A) amyloidosis (also termed secondary or reactive amyloidosis) results from increased production of serum amyloid A (SAA) protein, an acute phase reactant seen in chronic inflammatory conditions and infections. It primarily affects the kidneys, liver, and spleen.
Continue Learning with UWorld
Get the full Amyloidosis article plus rich visuals, real-world cases, and in-depth insights from medical experts, all available through the UWorld Medical Library.
Unlock Full AccessFigures
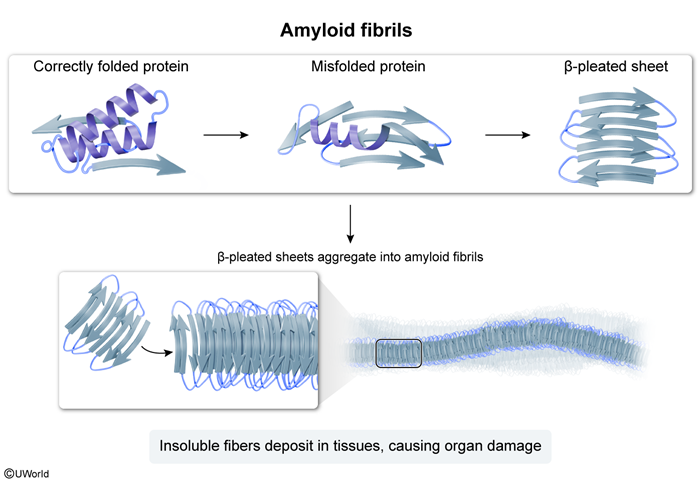
Figure 1
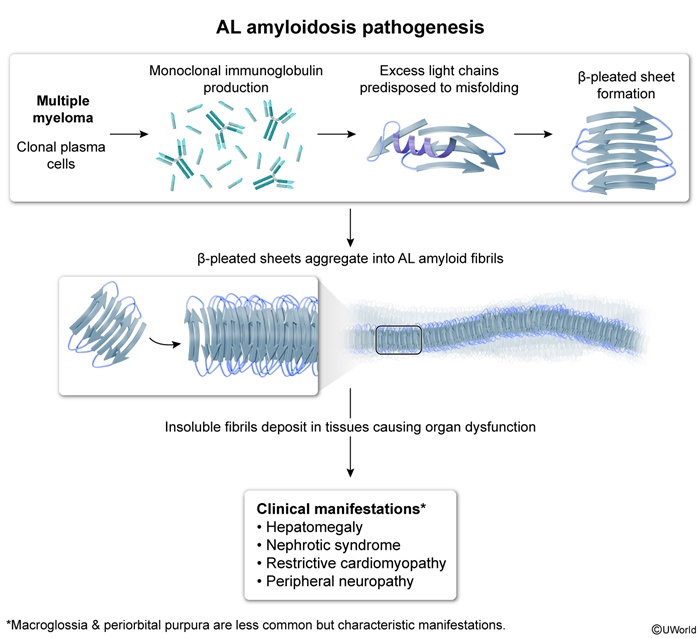
Figure 2
Images
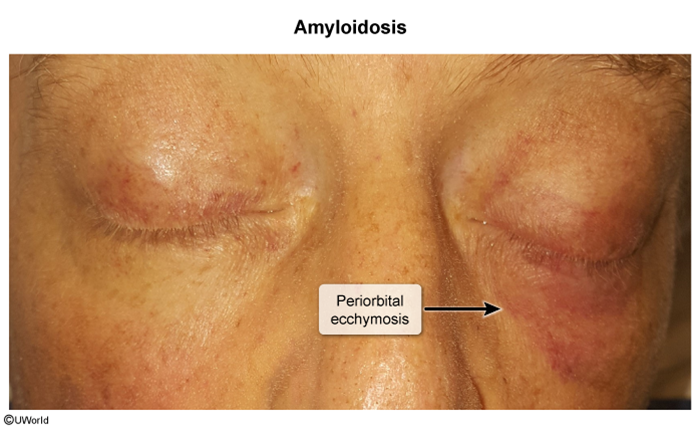
Image 1
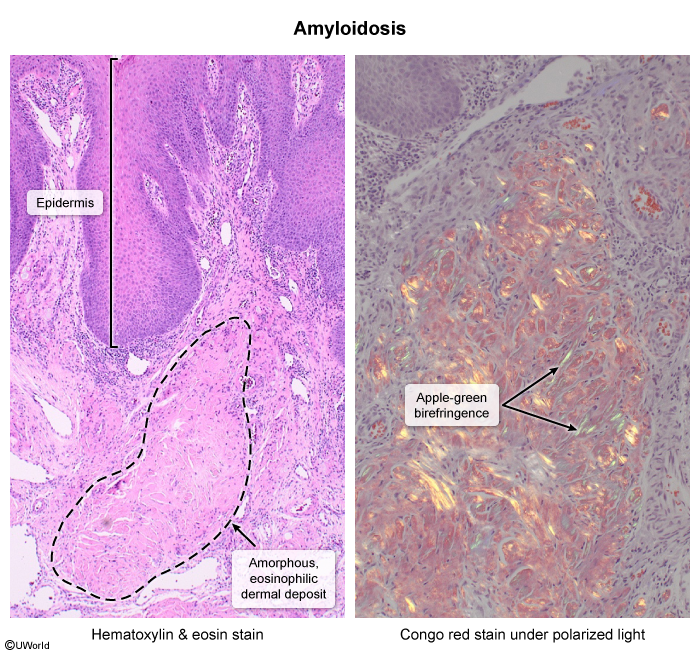
Image 2
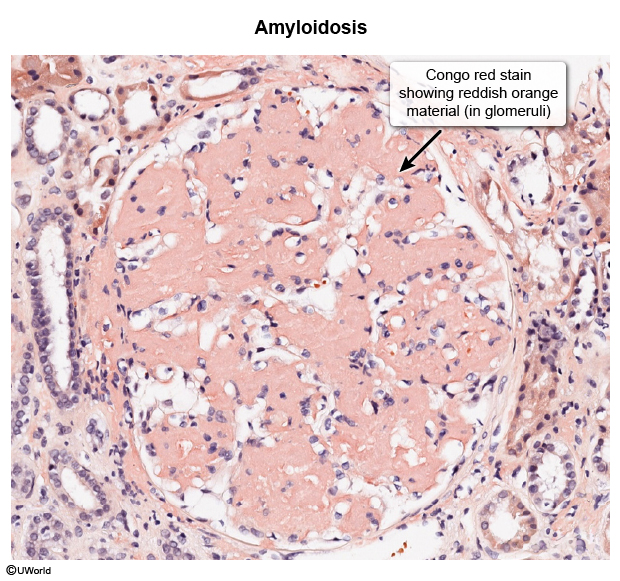
Image 3
Tables
Table 1
Medical Image
Medical Illustration