Wilson Disease
Article Sections
Introduction
Wilson disease (WD) is a rare autosomal recessive disorder characterized by excessive copper accumulation in the liver, brain, and other organs, leading to a wide range of hepatic, neurological, and psychiatric manifestations.
Pathophysiology
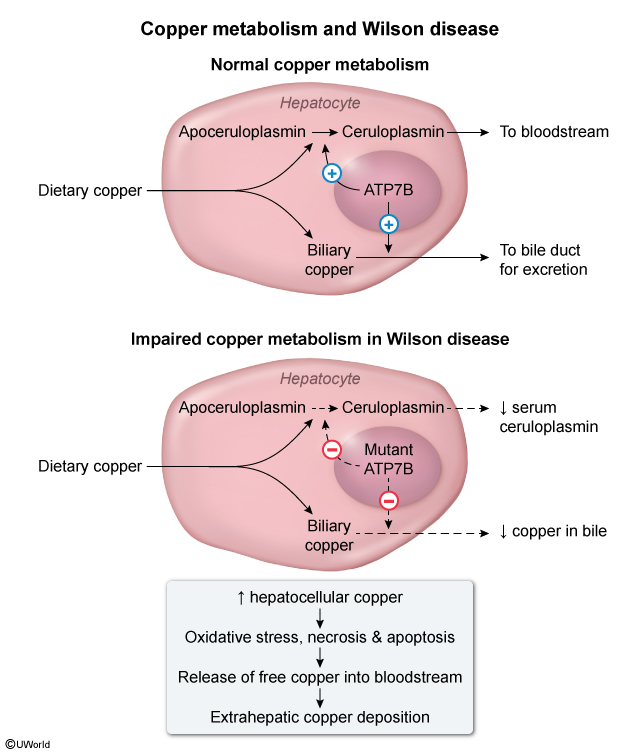
In general, dietary copper is absorbed in the stomach and duodenum and bound to circulating albumin. It is then transported to the liver and taken up by hepatocytes. The ATP7B protein in hepatocytes transfers excess copper into the bile canaliculi. This process is essential for biliary excretion of copper, which accounts for the majority of total body copper excretion.
In WD, a mutation of gene ATP7B hinders intracellular hepatocyte copper transport, causing decreased secretion of copper into the biliary system (Figure 1). The resultant increase in intracellular copper causes oxidative stress and apoptosis of hepatocytes, leading to liver dysfunction. Excess copper is then released into the bloodstream and deposited into extrahepatic tissues, including the CNS, kidney, and cornea. The basal ganglia is the primary site of deposition in the CNS and accounts for the movement abnormalities seen in WD. Because the basal ganglia, frontal cortex, and limbic system communicate, copper buildup also affects mood and cognition.
Continue Learning with UWorld
Get the full Wilson Disease article plus rich visuals, real-world cases, and in-depth insights from medical experts, all available through the UWorld Medical Library.
Unlock Full AccessFigures