Pancreatic Neuroendocrine Tumors (PanNETs)
Article Sections
Introduction
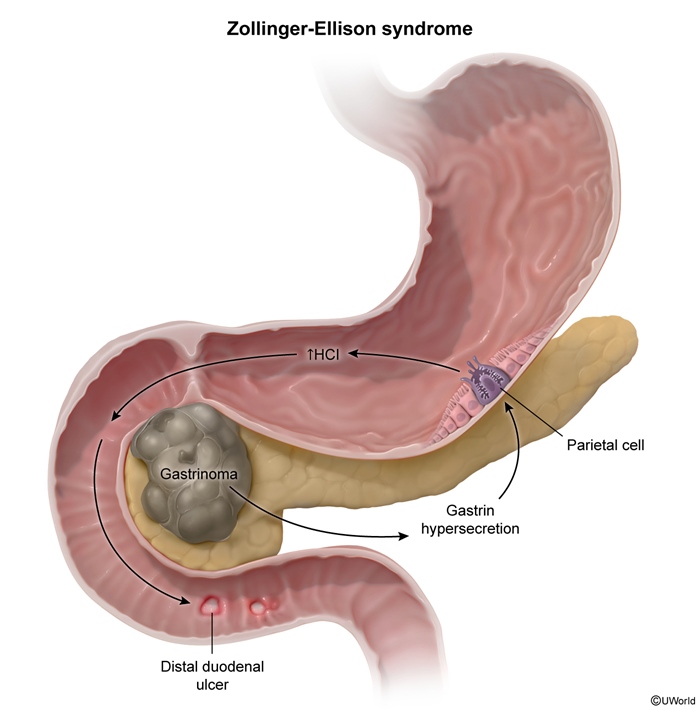
Pancreatic neuroendocrine tumors (PanNETs) are rare neoplasms derived from pancreatic islet cells. They are classified as functional PanNETs, which secrete peptide hormones and cause specific clinical syndromes (eg, insulinoma, gastrinoma, glucagonoma, VIPoma), and nonfunctional PanNETs.
Epidemiology and pathology
PanNETs are rare. Although many are sporadic, they can also be associated with hereditary syndromes, including:
- Multiple endocrine neoplasia 1 (~80%-100% of patients)
- Von Hippel-Lindau disease (~20% of patients)
- Neurofibromatosis 1 (~10% of patients)
- Tuberous sclerosis (~1% of patients)
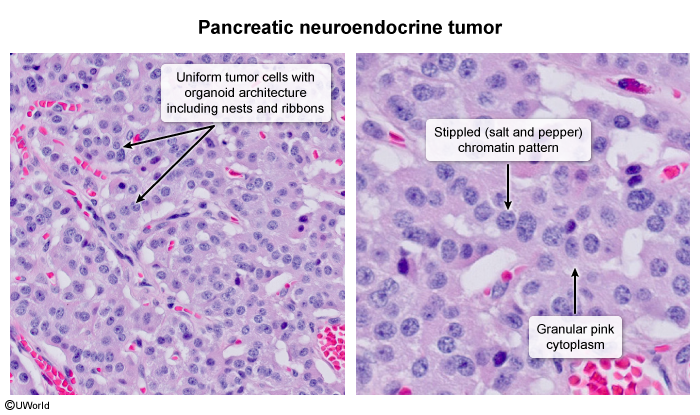
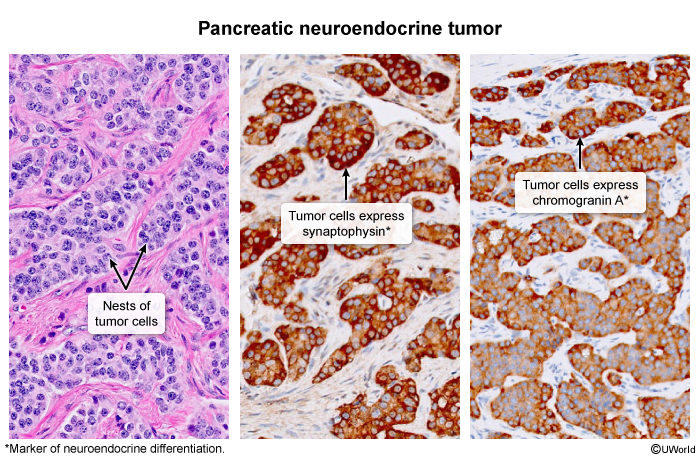
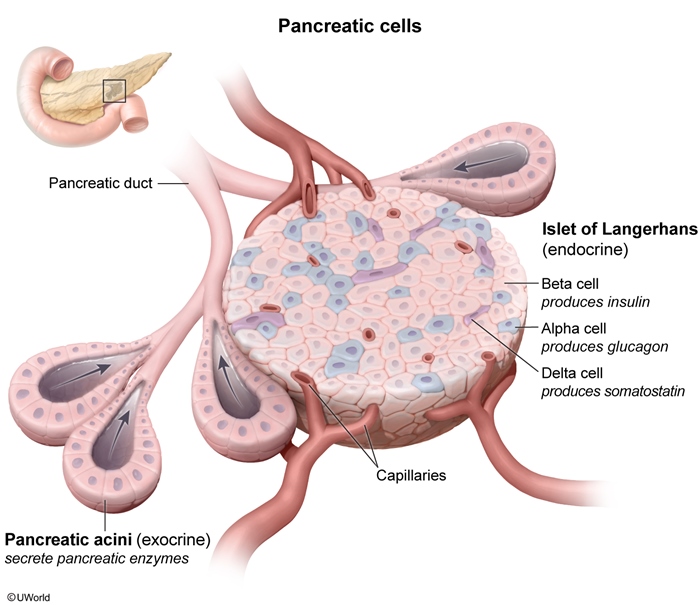
PanNETs originate from pancreatic islet cells (Figure 1). They can vary in size and location and can be solitary or multiple. Both functional and nonfunctional PanNETs are typically characterized by a well-circumscribed lesion composed of uniform tumor cells with an organoid architecture (eg, nested, glandular, ribbon, or pseudorosette arrangement) (
Continue Learning with UWorld
Get the full Pancreatic Neuroendocrine Tumors (PanNETs) article plus rich visuals, real-world cases, and in-depth insights from medical experts, all available through the UWorld Medical Library.
Unlock Full AccessFigures
Images