Hyper-IgE Syndrome
Article Sections
Introduction
Hyper-IgE syndrome is a primary immunodeficiency syndrome in which impaired Janus activated kinase–signal transducer and activator of transcription (JAK-STAT) signaling between T-helper cells and neutrophils prevents neutrophils from responding to infection. Patients have recurrent skin and sinopulmonary infections, eczema-like dermatitis, and characteristic physical features (eg, coarse facial features, hyperextensible joints).
Pathogenesis
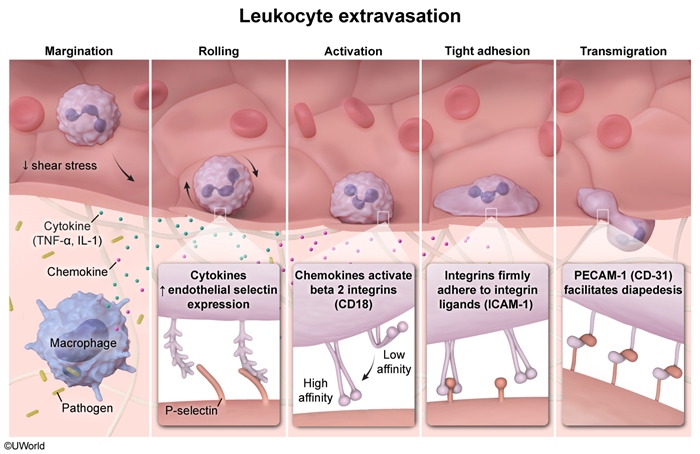
Neutrophils respond to infection through a multistep process that begins when circulating neutrophils extravasate and migrate to the site of infection (ie, chemotaxis) (Figure 1). A disruption in any step of this process can lead to persistent and/or recurrent infections, as seen in hyper-IgE syndrome.
Patients with hyper-IgE syndrome have a normal number of neutrophils capable of adhering, transmigrating, engulfing, and lysing pathogens. However, chemotaxis requires cytokine signaling. For example, T-helper cell type 17 (Th17) secretes IL-17 to induce neutrophil proliferation and chemotaxis in response to infection. Hyper-IgE syndrome is most commonly caused by an autosomal dominant mutation in the gene for STAT3, a cytoplasmic protein involved in the JAK-STAT pathway.
Continue Learning with UWorld
Get the full Hyper-IgE Syndrome article plus rich visuals, real-world cases, and in-depth insights from medical experts, all available through the UWorld Medical Library.
Unlock Full AccessFigures