Hypersensitivity Reactions
Article Sections
Introduction
Hypersensitivity reactions represent exaggerated immune responses to an antigen or an allergen. They explain certain allergic reactions but also other (eg, autoimmune) disease conditions. Four main types of hypersensitivity reactions are recognized (Gell and Coombs classification), based on underlying immune mechanisms and the time course of the reaction:
- Type I (immediate hypersensitivity) reactions are IgE-mediated, have rapid onset, and can be seen in atopic, allergic, and anaphylactic conditions.
- Type II (antibody-mediated cytotoxic hypersensitivity) reactions are characterized by IgG or IgM targeting host cells and interfering with their function or leading to damage, as seen in some autoimmune conditions (eg, autoimmune hemolytic anemia, antineutrophil cytoplasmic antibody [ANCA] vasculitis, Graves disease).
- Type III (immune complex hypersensitivity) reactions are characterized by immune complex (antibody-antigen) deposition in tissues, leading to inflammation (eg, serum sickness, lupus).
- Type IV (cell-mediated hypersensitivity) reactions are T cell–mediated, delayed-type (hours to days) reactions that underlie conditions such as allergic contact dermatitis and tuberculin skin test reactions.
The mnemonic ACID (type I: Allergic/atopic/anaphylactic; type II: Cytotoxic/cell-mediated; type III: Immune complex–mediated; type IV: Delayed) is sometimes used to recall these categories.
Type I hypersensitivity
An allergen is an antigen that promotes a robust immune response only in a subset of the population. Type I hypersensitivity reactions can occur locally (eg, cutaneous hives, allergic rhinitis [hay fever], bronchial asthma) or systemically (eg, anaphylaxis), depending on the allergen and its portal of entry into the body (video 1).
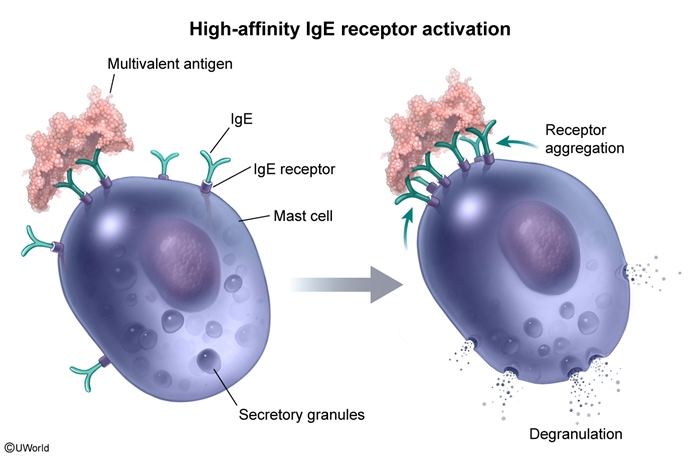
After first exposure to an allergen (eg, peanut proteins, beta-lactam antibiotic), antigen-specific IgE is produced by B cells (as a result of class switching) and binds to the high-affinity IgE receptor (FcεRI) on the surface of mast cells. This receptor normally binds the Fc portion of circulating IgE, coating the cell with various antigen-specific IgE molecules. With repeat exposure to a multivalent antigen, multiple IgE antibodies become cross-linked (Figure 1), resulting in aggregation of the FcεRI receptors on the mast cell surface. This clumping of receptors leads to the activation of nonreceptor tyrosine kinases, triggering an intracellular cascade that ultimately results in mast cell and basophil degranulation.
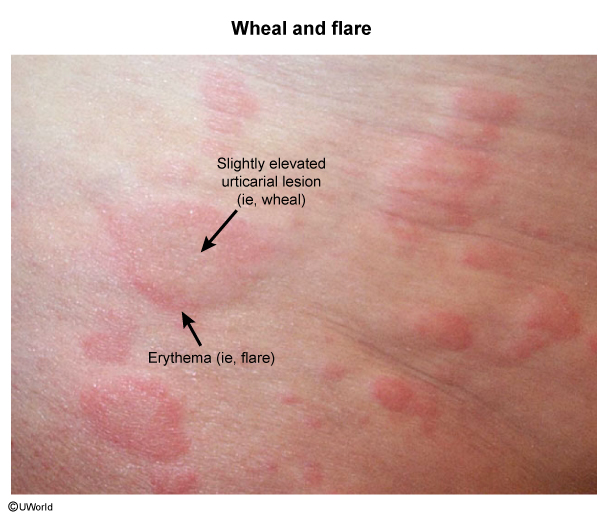
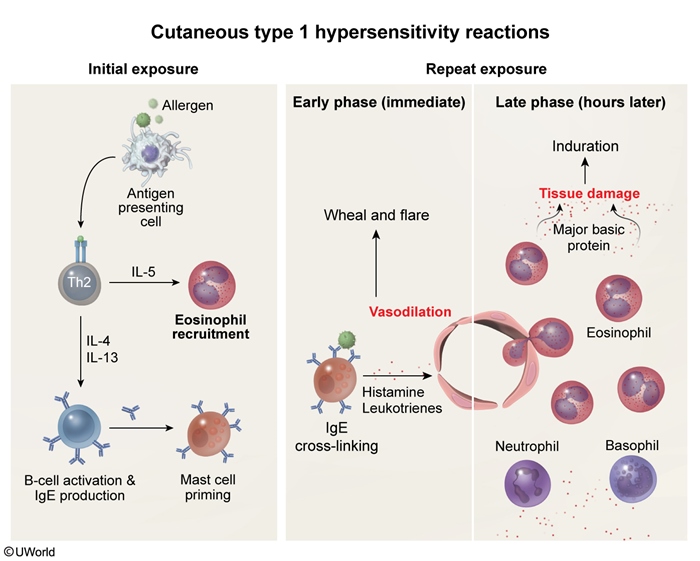
Systemic type I hypersensitivity reactions can lead to a potentially fatal, distributive form of shock (anaphylactic shock). Local reactions often have early-phase (eg, minutes after exposure) and late-phase responses (Figure 2). As an example, an early-phase cutaneous type I response could be superficial dermal edema and erythema (eg, wheal and flare reaction (Image 1)), which can progress to a more systemic response (eg, anaphylaxis). IgE also initiates the late phase of type I hypersensitivity reactions by stimulating type 2 helper T cells to release cytokines (eg, IL-5) that activate eosinophils; cationic proteins (eg, major basic protein, eosinophil peroxidase) released from eosinophils cause tissue damage. For example, a late-phase cutaneous type I response usually manifests as a palpable, indurated lesion 2-10 hours following the early-phase reaction.
Representative examplesType I hypersensitivity reactions are commonly thought of as:
- Allergic or atopic: "Atopy" refers to the predisposition to develop local immediate hypersensitivity reactions (eg, allergic rhinitis [hay fever], atopic dermatitis [eczema], bronchial asthma).
- Anaphylactic (eg, reaction to insect sting, peanuts, some medications)
Anaphylaxis, the classic manifestation of systemic type I hypersensitivity reaction, is likely if there is rapid symptom onset and any 1 of the following criteria:
- Skin/mucosa involvement (eg, hives, lip/tongue swelling) and either hypotension or respiratory distress
- Involvement of ≥2 organ systems after exposure to a likely allergen:
- Skin/mucosa (eg, hives, lip/tongue swelling)
- Respiratory (eg, wheezing, stridor, dyspnea)
- Cardiovascular (eg, hypotension, tachycardia, syncope)
- Gastrointestinal (eg, abdominal pain, vomiting, diarrhea)
- Hypotension after exposure to a known allergen
The diagnosis of type I hypersensitivity reactions is often made clinically. Complete blood count may show eosinophilia in some cases, and serum total and specific IgE levels might be elevated. Tryptase levels, which are specific for mast cells, can be elevated after anaphylactic reactions. Skin prick testing or specific IgE blood tests can help identify the culprit allergen. Genetic predisposition plays a role in these reactions, so family history may be positive.
Management- Avoidance: Eliminating the trigger allergen is the cornerstone of management.
- Antihistamines: Blocking the action of histamine alleviates related symptoms (eg, itching, hives, rhinorrhea).
- Mast cell stabilizers: Preventing mast cell degranulation can reduce the release of inflammatory mediators.
- Bronchodilators: In patients with respiratory symptoms, bronchodilators can relax airway smooth muscle and improve airflow.
- Epinephrine: In severe cases, especially anaphylaxis, immediate administration of epinephrine is crucial. Epinephrine is the only pharmacologic therapy that both decreases the release of inflammatory mediators from mast cells and addresses all other manifestations of anaphylaxis. It binds to adrenergic receptors (eg, beta-2 receptors) on the surface of mast cells, inhibiting mast cell degranulation. In addition, it counteracts existing inflammatory effects via widespread stimulation of alpha- and beta-adrenergic receptors:
- Alpha-1 receptor activation causes vasoconstriction and decreases vascular permeability, raising blood pressure and decreasing upper airway edema.
- Beta-1 receptor activation increases cardiac contractility and cardiac output, improving blood pressure and peripheral perfusion.
- Beta-2 receptor activation causes bronchodilation, relieving airflow obstruction.
In patients with severe reactions or in those who have an increased likelihood of exposure to hymenoptera venom (eg, pest control workers), allergen immunotherapy (AIT) is a disease-modifying treatment often recommended. With AIT, escalating doses of antigen are administered subcutaneously over several years. Progressive exposure induces immune tolerance via several immunologic mechanisms, including:
- Desensitization of mast cells and basophils (by triggering low-level release of intracellular granules) followed by decreased recruitment and activation of these cells (due to increased IL-10 and IgG4)
- Generation of blocking antibodies: AIT upregulates allergen-specific IgG4 antibodies. In contrast to IgG1 and IgG2, IgG4 cannot activate the complement cascade or serve as a costimulatory ligand for natural killer cells or phagocytes. Therefore, increased production of IgG4 helps bind and neutralize antigens without promoting additional immune activity.
- Shifting T-cell differentiation away from pathogenic Th2 subsets toward Th1 cells and regulatory T cells, increasing production of immunosuppressive cytokines (eg, IL-10, TGF-beta) and ligands (eg, CTLA-4, PD1) and reducing activation of mast cells and basophils.
- Decreased IgE production, which is caused by the shift from Th2 cell–driven responses to Th1-driven responses. Detectable reduction of serum IgE often takes years because IgE-producing plasma cells in bone marrow have long lifespans.
Type II hypersensitivity
Binding of preformed IgM or IgG antibodies to cell surface antigens can cause cell lysis, inflammation, or dysfunction. These reactions can develop hours to days after exposure to an antigen (eg, medications, transfused red blood cells), resulting in destruction of erythrocytes (causing fatigue, pallor), platelets (causing petechial bleeding), or leukocytes (causing fever, sepsis) (video 2).
The 3 main mechanisms include:
- Opsonization and phagocytosis: These occur through complement fixation or through (Fc receptor–mediated) antibody-dependent cellular cytotoxicity via neutrophils, monocytes, eosinophils, and natural killer cells. Examples include transfusion reactions, autoimmune hemolytic anemia, immune thrombocytopenic purpura, erythroblastosis fetalis, and certain drug reactions.
- Complement- and Fc receptor–mediated inflammation: Antibody deposition in extracellular tissue results in inflammation. Examples include certain forms of glomerulonephritis, Goodpasture syndrome (anti–glomerular basement membrane disease), vascular rejection (after transplant), and ANCA vasculitides.
- Antibody-mediated cellular dysfunction without injury or inflammation: Examples include myasthenia gravis, pemphigus vulgaris, and Graves disease (leading to antibody-mediated cell function stimulation).
Treatment depends on the cause and severity of the reaction and may involve immunosuppressive medications, intravenous immune globulin, or plasma exchange. Complications such as organ damage and bleeding complications from hemolytic anemia and thrombocytopenia should be managed as well.
Type III hypersensitivity
In type III hypersensitivity reactions, immune complex–mediated complement activation typically causes tissue damage in sites where immune complexes (ie, antibody-antigen) tend to deposit (eg, skin, joints, kidneys) (video 3). Manifestations tend to present days to weeks after initial antigen exposure. Antigens can be exogenous (eg, foreign protein, bacteria) or endogenous (eg, antigenic components of cells). Smaller immune complexes are more likely to be deposited in small vessels or sites where blood is filtered into other fluids, such as joints and kidneys. The underlying pathophysiology is as follows:
- Foreign proteins (eg, from a medication) are processed by antigen-presenting cells and displayed on major histocompatibility complex class II molecules (MHCII).
- Patrolling CD4+ cells bind the antigen-MHCII complex, undergo activation, and stimulate B cells to form plasma cells that secrete high-affinity IgG.
- The IgG binds the foreign proteins in the medication, thereby generating immune complexes.
When large quantities of immune complexes are rapidly formed, they aggregate and deposit in the skin and joints. Deposited immune complexes activate the classical complement cascade. Although this generates the opsonizing protein C3b, which allows the complex to be cleared by phagocytic cells using the C3b receptor, it also generates the highly inflammatory anaphylatoxins C3a and C5a.
C3a and C5a are potent chemoattractants that dramatically increase phagocyte recruitment (eg, neutrophils, macrophages) to areas of complex deposition due to effects on the vasculature (eg, vasodilation, increased permeability) and immune cells (eg, increased adhesion molecule expression). This leads to development of local (eg, skin rash, joint pain/effusion, neutrophilic joint effusion) and systemic (eg, high fever, elevated erythrocyte sedimentation rate) inflammatory manifestations. Immune complex injury leads to necrotizing vasculitis (eg, fibrinoid necrosis), and the complexes can sometimes be seen on microscopy (eg, in glomerulonephritis).
Representative examplesSimilar to type I hypersensitivity, some type III hypersensitivity reactions lead to generalized/systemic diseases whereas others cause localized disease, depending on the site of immune complex deposition.
Serum sickness is a classic example of generalized/systemic type III hypersensitivity reaction. In the 1900s, patients injected with serum from horses immunized with diphtheria toxin developed rash, arthritis, and fever; this condition was due to patient antibodies to horse serum proteins forming complexes with the proteins in the injected serum. In present-day settings, serum sickness can occur following administration of antigenic heterologous proteins such as chimeric monoclonal antibodies (eg, rituximab and infliximab) or nonhuman immunoglobulins (eg, venom antitoxins). A serum sickness–like reaction is also associated with the use of certain nonprotein drugs (eg, penicillin, cefaclor, trimethoprim-sulfamethoxazole) (Table 1). Continuous exposure to an antigen can cause chronic immune deposition disease; a representative example is systemic lupus erythematosus (which can cause nephritis and arthritis, among other conditions).
Local type III hypersensitivity is illustrated by the Arthus reaction. This local, immune complex–mediated area of tissue necrosis in the skin is usually associated with intradermal injection of a vaccine. Similar to type I hypersensitivity reactions, Arthus reaction can cause localized skin swelling, but this occurs within hours of an exposure (vs immediately with type I hypersensitivity reaction); in addition, it can progress to hemorrhage and ulceration.
Type IV hypersensitivity
In type IV (delayed) hypersensitivity reactions, CD4+ T cells release cytokines (eg, interferon gamma) that promote macrophage- and T cell–mediated tissue damage. When reexposed to an antigen, previously sensitized T lymphocytes (eg, memory T cells) proliferate and release inflammatory cytokines that promote cell-mediated cytotoxicity (CD8+ T cells) and/or macrophage recruitment and activation. The resulting tissue damage and swelling is typically evident 24-48 hours after exposure, once sufficient cellular proliferation has occurred.
Representative examplesAllergic contact dermatitis (eg, poison ivy), reactive skin testing (eg, tuberculin skin test, Candida extract skin reaction), and granulomatous inflammation are examples of type IV (delayed type) hypersensitivity reactions.
Allergic contact dermatitis
Allergic contact dermatitis (eg, urushiol in poison ivy) occurs in 2 phases:
- In the sensitization phase, cutaneous Langerhans cells take up haptens (ie, allergens) and present them to naive CD4+ and CD8+ T cells in the regional lymph nodes, resulting in clonal expansion of hapten-sensitive T cells. This phase takes 10-14 days and does not result in cutaneous lesions.
- On reexposure to the hapten, cutaneous antigen-presenting cells present the hapten to sensitized T cells recruited to the skin. When activated, these T cells mediate tissue damage that manifests as pruritic erythema, vesicles, and/or bullae. This elicitation phase occurs 2-3 days following reexposure to the hapten.
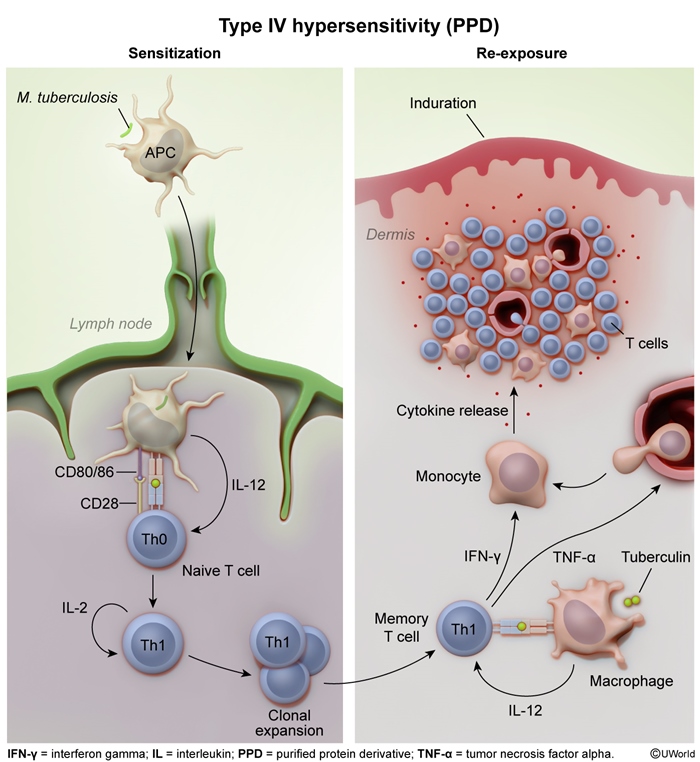
Tuberculin skin testing (Figure 3)
Tuberculin skin testing (eg, purified protein derivative) introduces purified proteins from Mycobacterium tuberculosis into the dermis. Because M tuberculosis is primarily countered by the cell-mediated immune response, previously infected patients have primed, antigen-specific CD4+ T lymphocytes that rapidly replicate and mature in response to tuberculin antigen reexposure.
T-lymphocyte activation is a 2-step process:
- T lymphocytes with specific T-cell receptors recognize antigens presented on MHCII on the surface of dendritic cells (eg, antigen-presenting cells [APCs]).
- A costimulatory interaction between CD28 on the T lymphocyte and CD80/86 on the dendritic cell allows for activation.
Activated CD4+ T lymphocytes release inflammatory cytokines (eg, IL-1, IL-6, TNF alpha) that stimulate/recruit other immune cells and increase vascular permeability, forming an indurated wheal of inflammation following tuberculin skin testing exposure. Tuberculin reactions appear 24-72 hours after antigen exposure due to delays between initial antigen processing by APCs, T-cell activation, and amplification of the cellular response.
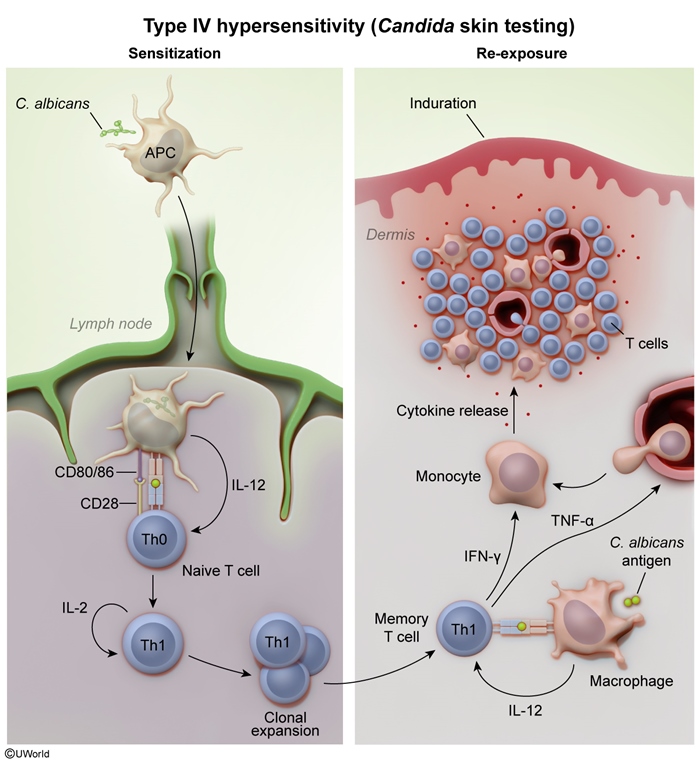
Candida extract skin reaction (Figure 4)
Intradermal injection of Candida antigens triggers a type IV hypersensitivity reaction, which allows for evaluation of the cell-mediated immune response. Injected Candida antigens are taken up by cutaneous APCs, which display the antigens on MHCII molecules to naive CD4+ cells. These cells are then induced to differentiate into Th1 cells that secrete tumor necrosis factor and interferon-gamma, which recruit and activate macrophages. The macrophages phagocytose Candida antigens and secrete additional inflammatory mediators, producing a cutaneous inflammatory response that appears as an indurated wheal within 24-48 hours. Because exposure to Candida is ubiquitous, all healthy patients have memory T cells against Candida. Failure to generate an appropriate cutaneous response indicates that the cell-mediated immune response is impaired (ie, T-cell anergy).
Summary
The 4 main hypersensitivity reactions have distinct immunological mechanisms (Table 2). Type I reactions, mediated by IgE and mast cells, cause immediate allergic responses (eg, anaphylaxis). Type II reactions involve binding of preformed IgM or IgG antibodies to cell surface antigens, which can cause cell lysis, inflammation, and dysfunction, potentially leading to autoimmune diseases. Type III reactions arise from immune complex deposition and complement activation, resulting in conditions such as serum sickness. Type IV reactions are cell-mediated by T cells and take 24-48 hours to develop, as seen in allergic contact dermatitis and tuberculin skin tests. The mnemonic ACID (type I: Allergic/atopic/anaphylactic; type II: Cytotoxic/cell-mediated; type III: Immune complex–mediated; type IV: Delayed) can be helpful.
Continue Learning with UWorld
Get the full Hypersensitivity Reactions article plus rich visuals, real-world cases, and in-depth insights from medical experts, all available through the UWorld Medical Library.
Figures
Images